Accounts of a microbial hunter in France:
Murphy’s Law with a twist - Whatever can go wrong, will go wrong…however, valuable discoveries can be made
“It’s important to grow comfortable with the unknown and unexpected. As these are teachable moments where valuable information and skills can be acquired and refined”

Microbes involved in biologically removing nitrate are ubiquitous to terrestrial and aquatic ecosystems. Perhaps we can have a look for our microbial counterparts at the Calanques in Marseille, France!
If I could summarize these previous weeks, it would go according to Murphy’s law “whatever can go wrong, will go wrong.” However, I realize that this is nature of research - our plans rarely go as anticipated. It’s important to grow comfortable with the unknown and unexpected. As these are teachable moments where valuable information and skills can be acquired and refined. In fact, some of the greatest scientific discoveries were made when the unexpected occurred - penicillin, the pacemaker, and post it notes (I am biased towards this one).
In this blog post, I share my experience with navigating through a particular challenge that halted our analysis. This piece also includes a reflection of my experience with troubleshooting different problems.
Prior to beginning, I extend a warm welcome to you all. You all are my fellow microbial hunters, who are joining this journey to search for microbes involved in treating nitrate contaminated drinking water. For my newest microbial hunters, I would like to direct you to the first blog post that provides information on the motivation of this project and background. This piece is a resource that sets the stage for the subsequent posts of this series.
Let’s proceed with hunting for our microbial counterparts!
Navigating through the unexpected
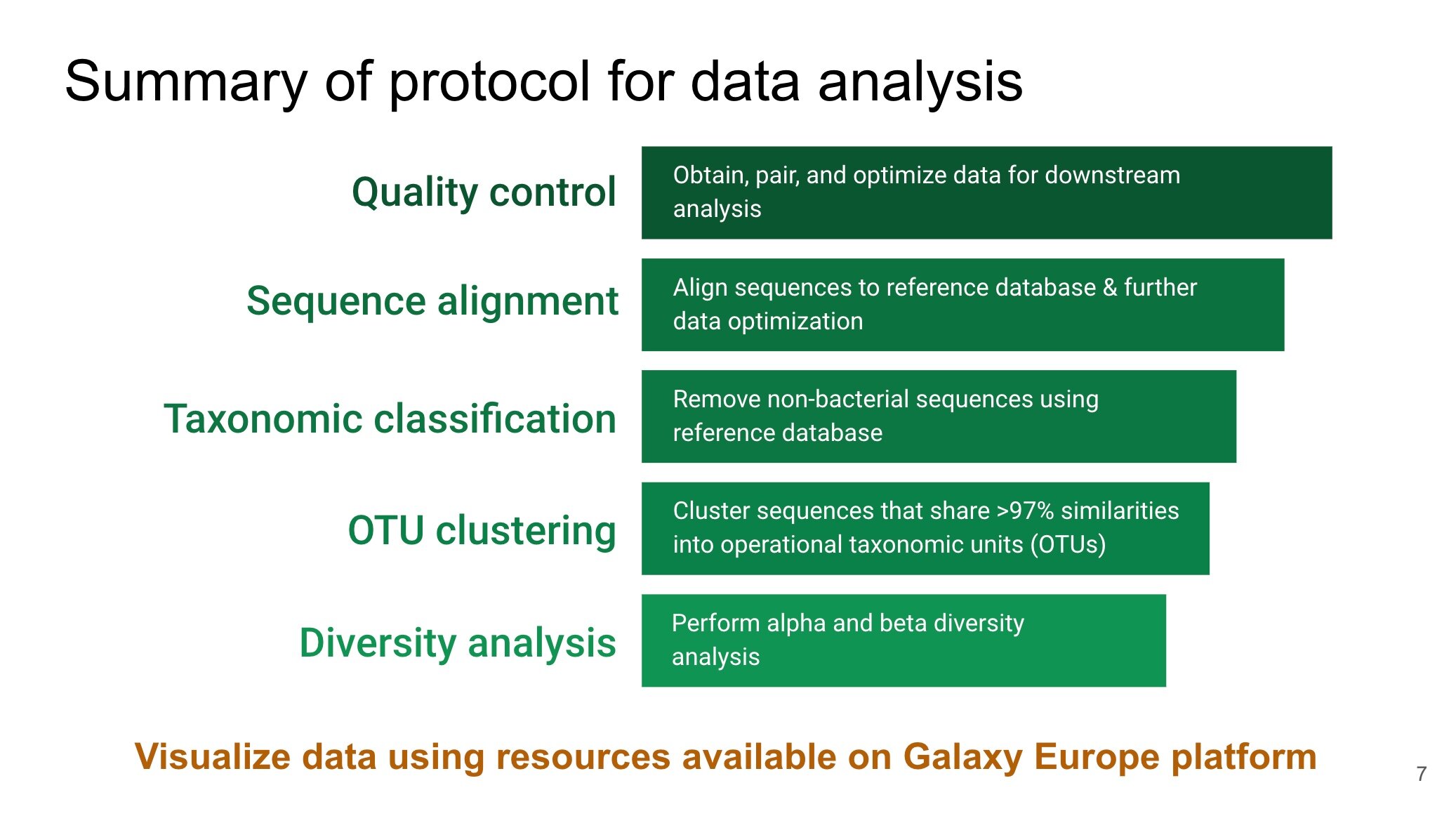
We are using the Galaxy 16S Microbial Analysis tutorial and Mothur MiSeq SOP as guides to process and analyze our next generation sequencing (NGS) big data. This big data represents the microbes within samples collected from our engineered treatment systems. We have decided to use the workflows of these tutorials as a guide for two reasons: ( 1 ) Both tutorials provide hands-on experience to examine big data associated with the 16S gene, which is frequently used for identifying microbes and other organisms. The data included in the tutorials can be used as a positive control. A positive control produces an expected outcome and can assists to some extent with troubleshooting and for making comparisons with the big data from our studies; ( 2 ) Other researchers have mentioned using the workflow of these tutorials in the methods section of their peer reviewed publications. This substantiates that these tutorials have been accepted by our research community.

We are using the Galaxy 16S Microbial Analysis tutorial as a guide to process and analyze the next generation sequencing big data that represents the microbes collected from our engineered treatment systems.
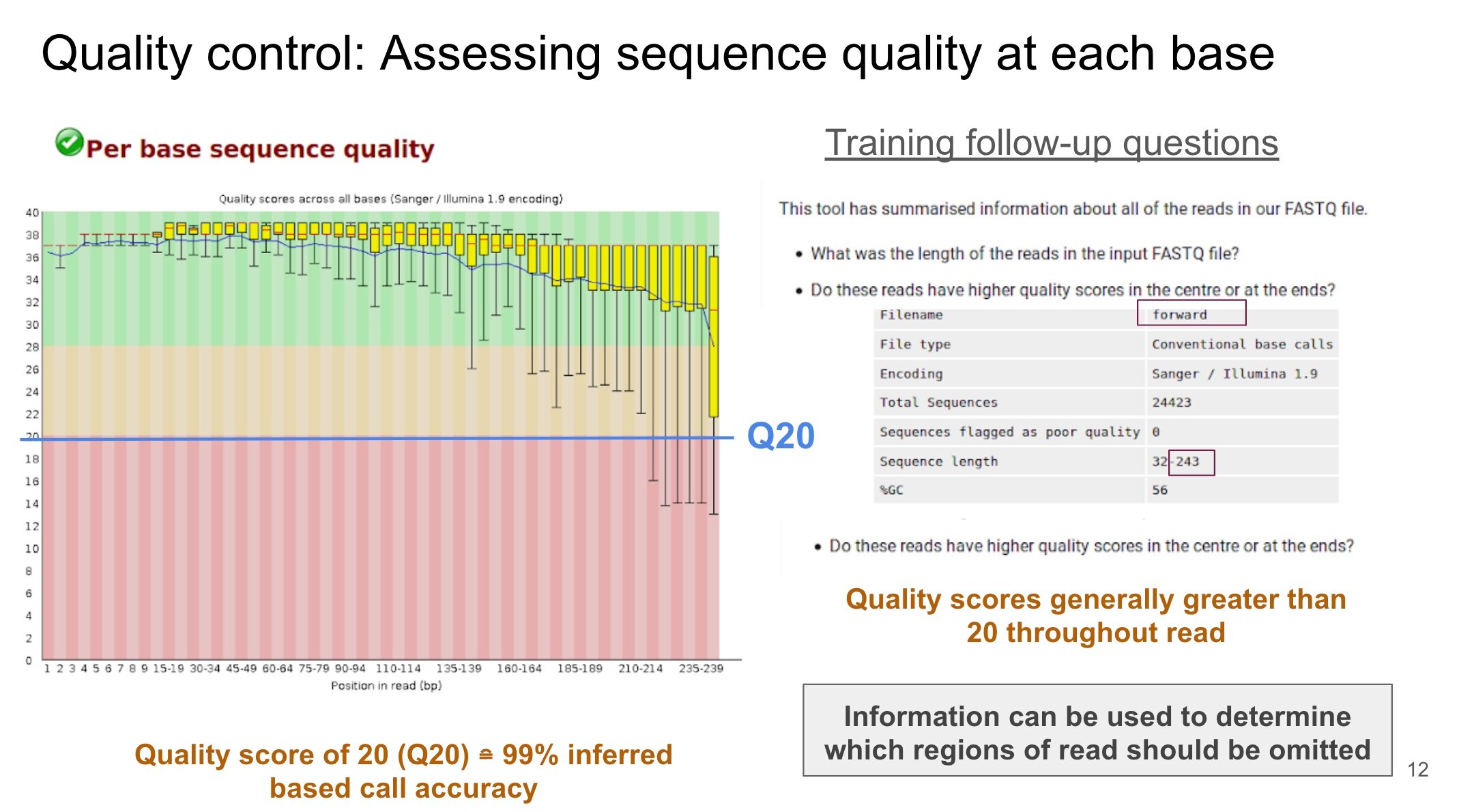
The beginning of the tutorial presents an optional QC control step, which can be used to remove low quality portions of data. Although the tutorial does not include this step, there is a separate online resource on the Galaxy website to acquire practice with performing it. In our case, we used the Trimmomatic QC tool since it has been applied by other researchers in our field of water research.
It’s important to note that the big data produced by NGS represents individual segment of the organisms that are present within a sample. This is based on how the samples were prepared for NGS. These individual segments must be combined or “glued together” in order to gain useful information about microbial community composition. Therefore, after the QC step, we attempted to assemble our big data into larger segments known as contigs. However, we were unsuccessful with connecting these two steps.

An example of big data from the Galaxy 16S Microbial Analysis tutorial, which has been combined into segments known as contigs.
The Galaxy application has a Gitter page where community members can seek assistance. Galaxy technical advisors respond relatively quickly to post added to the Gitter chatroom.
I used a combination of approaches in attempts to resolve this challenge. I began with creating a post on the Galaxy Help website, where administrators and community members can provide assistance and feedback. Shortly after, I discovered the Galaxy Gitter page. This page is a chatroom where community members can post questions and/or concerns. One advantage of this resource is that users can chat directly with Galaxy technical advisors and receive a response relatively quickly. Both of these resources suggested revisiting my workflow in Galaxy to ensure that input files were included. Furthermore, I received feedback to check the information icon on the error output file to access the stdout page. This page provides the error code, a description of the error, and advice for troubleshooting. If necessary, the error code can be included in a bug report to give additional context to a Galaxy administrator.
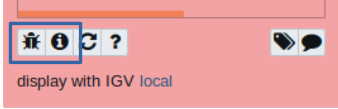
Error output files contain the bug report and information icons for seeking assistance and troubleshooting, respectively.
Following the advice from the Galaxy community, I accessed the stdout page. I learned that the underlying issue was related to the naming system, or nomenclature, used to identify the big data for each sample. Interestingly, I realized that certain characters ( . and - ) have specialized functions in the underlying program that Galaxy uses to analyze the microbial community (i.e., Mothur). As a result, using these characters to name the big data representing a sample can interfere with a tool and generate an error output. I proceeded with modifying the sample nomenclature and the problem was resolved. The QC step using Trimmomatic could be connected to the first step of the Galaxy 16S Microbial Analysis tutorial - creating contigs.

Specific characters ( e.g., _ or . ) are sensitive in Mothur and must be omitted to prevent errors from occurring.
Lessons learned
“Instead of avoiding the errors that I encounter, I embrace and see them in a new light - they are opportunities to think critically and creatively. In the end, I am improving my problem solving skills, which can be applied to both my research and life. ”

Error outputs that I received during another halt that I encountered.
Initially, I had anticipated that the analysis of the big data would go smoothy. This was because other researchers have demonstrated their success with following the Galaxy 16S Microbial Analysis tutorial and MiSeq SOP workflows. However, I am grateful for this halt (and the others) occurred because I was able to discover valuable information.
The experience enabled me to learn about different resources that are available to Galaxy members. Now, that I am aware of them, I can visit them in the future to seek assistance. This experience also allowed me to become familiar with the stdout file, which I have used on other occasions for troubleshooting errors.
Furthermore, other halts have allowed me to realize that there is value in learning how the Mothur program performs. The Galaxy application provides operational simplicity for analyzing NGS big data. The user selects their desired parameters and Galaxy performs the computational work needed to operate Mothur. While the workflows of Galaxy 16S Microbial Analysis tutorial and MiSeq SOP can be used as guides, suitable parameters should be selected for a particular dataset. This is important for obtaining results that provide a close representation of a sample’s microbial community composition. Therefore, one must understand the purpose that each parameter serves. Another benefit of possessing this knowledge is that it can assists in resolving errors and refining existing workflows.
Ultimately, the challenges that I have experienced thus far have helped in building my confidence as a researcher. I used to be reluctant to troubleshoot my research out of fear that I might make the problem worst or I simply didn’t know where to begin. However, I have become resourceful in my efforts and no longer afraid of tackling an error output. Instead of avoiding the errors that I encounter, I embrace and see them in a new light - they are opportunities to think critically and creatively. In the end, I am improving my problem solving skills, which can be applied to both my research and life.
Acknowledgements
I would like to thank the Chateaubriand Fellowship, Alfred P. Sloan Foundation, and McKnight Doctoral Fellowship for supporting this project and encouraging opportunities for graduate students to pursue research abroad!
Thank you for your time and I hope that you were able to learn something new from this post. For any additional questions or comments feel free to leave your responses below!